Virtual Screening of New azo Coumarin Derivatives as Possible Alkaline Phosphatase Inhibitors
DOI:
https://doi.org/10.5530/ctbp.2024.3.38Keywords:
Alkaline phosphatase, coumarin, ADMET, Drug-likeness, Molecular dockingAbstract
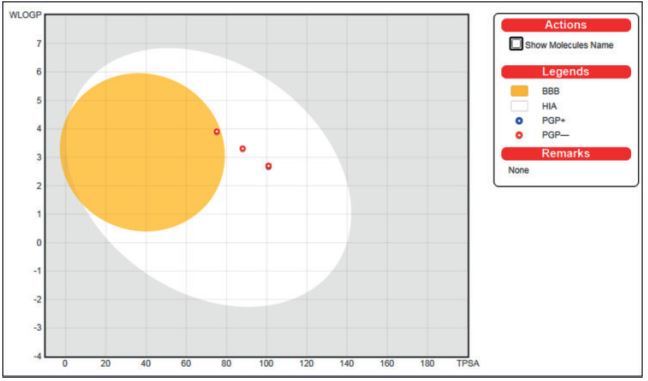
To develop new alkaline phosphatase inhibitors, a series of new azo coumarin derivatives were designed by using computer aided drug designing and virtually assessed using online platforms. At first, the compounds were screened for ADMET, physicochemical properties, drug-likeness, toxicity studies and target prediction using pKCSM, SwissADME, SwissTargetPrediction and ProTox-II tools. The predictions were supported by in silico molecular docking with alkaline phosphatase enzyme using CB-Dock2 molecular docking tool. The compounds possessed good ADMET and physicochemical properties, drug-likeness and devoid of any immunotoxicity and cytotoxicity. The evaluated binding energy values reveal that all compounds fit favorably into the alkaline phosphatase active site displaying hydrogen bonding with different amino acid residues of the target protein and could be good scaffolds for designing new alkaline phosphatase inhibitors. These results collectively framed the way for the development of new azo coumarin derivatives as possible alkaline phosphates inhibitors.